
{kind=link}
La ectopia cordis es una enfermedad rara que se define por una posición anormal del corazón fuera del tórax. Asociada a defectos del pericardio parietal, diafragma, esternón y, en la mayoría de los casos, cardiopatía.
Aspectos téoricos de la ectopia cordis
La nominación de ectopia cordis fue propuesta por primera vez por Abott en 18981, si bien, pacientes con defectos similares habían sido descritos en décadas anteriores con otras nominaciones.
Byron clasificó la ectopia cordis en 4 grupos: cervical, torácica, toracoabdominal y abdominal. En este último grupo se incluye a pacientes con afección de la línea media abdominal (onfalocele) que cumplen las características definitorias de la enfermedad.
En 1958, Cantrell, publicó un síndrome con 5 defectos: anomalías de pared torácica, abdominal, diafragma, pericardio y corazón.
CASO 1
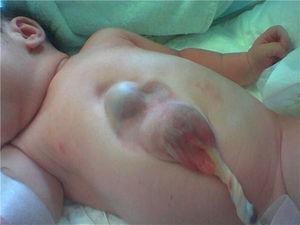
Presentamos un caso de ectopia cordis en una recién nacida pretérmino (36 semanas), mediante parto vaginal eutócico, que cursó sin complicaciones.
Se trata de un neonato con un defecto en pared toracoabdominal. En la región inferior esternal protuye la punta cardiaca cubierta por tejido cutáneo y una hernia supraumbilical.
Se realiza un ecocardiograma que descarta cardiopatía congénita y resonancia que confirma el diagnóstico de ectopia cordis toracoabdominal. Con defecto en línea media de la pared torácica anterior baja, herniación del apex, pequeño defecto diafragmático en la inserción con esternón y defecto supraumbilical por el que se hernia parcialmente el lóbulo hepático derecho.
A los 6 días de vida se repara quirúrgicamente, se coloca el corazón en posición intratorácica, se liga la vena umbilical y se liberan las adherencias a pared de lóbulo hepático derecho. La evolución es satisfactoria.
Ectopia cordis es un defecto raro, con una incidencia de 5,5–7,9 casos por millón de nacidos vivos, en que se expone el corazón, total o parcialmente, en el tórax.
Se clasifican en cervical, torácico, toracoabdominal y abdominal. Más del 80% de los fetos afectados asocian cardiopatía (generalmente las formas toracoabdominal y torácica). Como comunicación interventricular, tetralogía de Fallot, divertículo ventricular izquierdo, doble tracto de salida ventricular derecho e hipoplasia pulmonar. Representa el 0,1% de las cardiopatías congénitas. Es un defecto durante la embriogénesis, en que falla la fusión de los pliegues laterales en el área torácica durante la 6.ª semana posmenstrual. Este es uno de los pocos casos que no asocia cardiopatía.
CASO 2
Recién nacido de 42 semanas de gestación y peso natal de 2.900 g. En el nacimiento destacaban los siguientes datos: test de Apgar 7 y 9 (5 min), cianosis de piel y mucosas, polipnea con tiraje subcostal. Así como onfalocele con esternón abierto y corazón en la zona medial.
En la radiografía de tórax se observaba ausencia del diafragma, con las vísceras abdominales en el tórax (ectopia cordis toracoabdominal incompleta). El paciente fue intervenido, y falleció a las 24 h. En la necropsia cardíaca destacaba un drenaje de la vena cava superior e inferior en la aurícula derecha y de las venas pulmonares en la aurícula izquierda, atresia mitral, comunicación interventricular perimembranosa de salida, y emergencia de la arteria pulmonar y de la arteria aorta del ventrículo derecho.
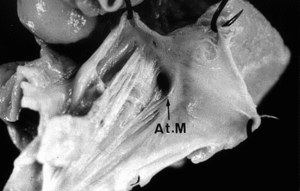
Además, se observaba estenosis valvular pulmonar e hipoplasia ligera del ventrículo izquierdo. Las arterias coronarias nacían de los senos anteriores e izquierdo.
Notas relacionadas:
¡La ciencia avanza! Hong Kong crea una prueba para DETECTAR COVID-19 prolongado
10 errores que cometen los médicos inteligentes ¡EVÍTALOS!
NOM mexicana oficial para el control de la hipertensión arterial